Continuous-Flow Measurement of Radioactivity in HPLC Eluates
Edward Rapkin, Ph.D.
In 1959 when Lyle Packard and I undertook development of what became the first commercial flow-through beta-activity monitor for liquid chromatography eluates (1,2), neither of us appreciated the full potential such an instrument offered. Rather, we thought that perhaps we had a useful complement to the Stein-Moore amino acid analyzers of that day for amino acids were, in truth, the only class of compounds that we could measure with confidence. Even so, almost anything one could think of, including amino acids, was better measured by collecting discrete fractions and counting them in a liquid scintillation counter; the liquid scintillation counter was less costly, less problematic, and more accurate. But, the picture has changed and now flow-through measurement is clearly the method of choice for following radioactivity elution from liquid chromatography columns. It is instructive to consider how this change, which had as much to do with peripheral factors as with measurement methods, came about.
Early systems
In 1958, with scintillator expensive and having interest in sample recovery, Steinberg (3) proposed that aqueous solutions containing beta-activity might be counted in vials in a liquid scintillation counter using suspended crystalline anthracene as a scintillator and a trace of detergent as a wetting agent. With the measurement completed, the anthracene could be filtered off for reuse and the filtrate processed as required. Reflecting beta-particle energy differences, C-14 (Emax = 156 kev) counting efficiencies (15-20%) were possibly acceptable but the best liquid scintillation counter gave no more than 0.5% H-3 (Emax = 18 kev) efficiency which was not.
Steinberg's method never aroused much interest; the added work was a bother, scintillator became more readily available, more sensitive liquid scintillation counters allowed samples to be divided with only the part that was counted being lost, and there were questions of how reproducible a suspension could be made. Also, anthracene is soluble in organic solvents thereby eliminating many possibilities as to both labeled compounds and solvent systems.
About the same time (1957-1959) Schramm (4,5,6) and Funt (7) constructed flow-through monitors based on single tube liquid scintillation counters in which solutions containing activity were passed through grooves in plastic scintillator blocks or through tubing made of plastic scintillator. C-14 could be measured. However, with low surface-to-volume ratio and with the energy of H-3 beta-particles so low as to be absorbed in a fraction of a millimeter of water, counting efficiencies for H-3 were low or non-existent. With single-tube counters, backgrounds were high; there was little incentive to pursue the method.
Our thought was to combine Steinberg's use of anthracene with Schramm and Funt's continuous measurement and to construct a commercial device using then state-of-the-art detection and electronics. We began with coil cells of polyethylene or Teflon but they were not easily packed and we turned to straight heavy-walled Kel-F tubing; coincidence-type electronics with water-cooled photomultipliers gave acceptably low backgrounds; use of optical light guides and closely-spaced photomultipliers made for improved counting efficiencies. But, even so, we had a system of only limited utility.
Anthracene, while an excellent scintillator, is attacked by organic solvents. Worse, it forms complexes with many of the substances that one might wish to count; the contaminated cell background was often unacceptable and the cells resisted decontamination. Still another consideration was that data treatment is impossible unless eluate flow is known and most liquid chromatography of that time depended upon gravity flow.
By 1960, chromatographic methods for amino acid analysis had become routine and commercial systems were built specifically for this purpose. Identification of amino acids by retention time required constant eluate flows which were achieved with metering pumps. Labeled amino acids did not adhere to anthracene nor did the buffers used for column elution have deleterious affects. Radio-labeled amino acid analysis seemed ideal for our first flow-through monitor. Experience proved otherwise.
H-3 counting efficiency at less than 1% was still too low; the flow-through monitor was essentially a C-14 counting device. Runs were long -- 6 hours or more -- thereby tying up expensive equipment for long periods. Data was collected with a strip-chart recorder; digital data was not easily had. Straight-tube cells were a poor compromise; too large a diameter meant peak spreading and photon loss within the cell while small diameters meant unacceptably short counting times. Though C-14 amino acid results were adequate and the convenience of real-time measurement was recognized, the method fell into disuse.
The principal reason for this went beyond those just cited and was a subtle consequence of the long chromatography runs. Assuming no cell contamination and therefore relatively constant background, when a peak is broad the total background collected over the peak width is much greater than would be true had that same peak been narrower and sharper, i.e., the signal to noise ratio is much less for the broader peak than for a better defined one having the same total activity. As a consequence, sensitivity of the continuous-flow system is hostage to the quality of the chromatography. The same flow system that is of no value with a lengthy chromatography run may provide worthwhile results as the run is shortened and the resolution improved.
Why not combine the chromatography eluate with liquid scintillator and pass the mixture through a coil placed in the counting chamber of a liquid scintillation counter? H-3 and C-14 efficiencies might reach 15-25% and 60-70% respectively, figures routinely achieved with the liquid scintillation counter. It seems an obvious thing to do and, in fact, it was first reported in 1962 (8). But, this was by no means a panacea. It required constant eluate flow so that the counting mixture had a constant composition and, more importantly, with the slow chromatography of those days, scintillator consumption was enormous and costly.
HPLC turns a curiosity into a useful method
What brought back continuous-flow measurement was the advent of HPLC (Figures 1 and 2). HPLC meant relatively short reproducible chromatography runs made with constant volume pumps; the continuous-flow monitor previously available for only 1-2 runs per day, could now be used for 1-2 runs per hour. And, with faster chromatography, runs that barely showed measurable radioactivity now gave useful results. In terms of throughput, the continuous-flow monitor was instantly an appreciably less expensive device than the liquid scintillation counter. Not only was first cost less, but so was operating cost.
For continuous monitoring using liquid scintillator, the incoming eluate from an HPLC column is mixed with scintillator at a tee and the mixture passed through the cell for measurement. Usual HPLC eluate flows are 0.5-2 mL/min and liquid scintillator:eluate ratios are from 3:1 to possibly, though rarely, up to 8:1. Scintillator consumption for a one-hour chromatography run might be as high as 1 l but is normally not even a third of that. Contrast that with fraction collection where a 60-minute chromatography run might give rise to 120 fractions each requiring 10 mL of liquid scintillator as well as a counting vial; supply cost is at least double and more often four or five times that for continuous-flow measurement (Table 1). Then, each vial might occupy a liquid scintillation counter for from 1 to 10 minutes for a total counting time of 120-1200 minutes.
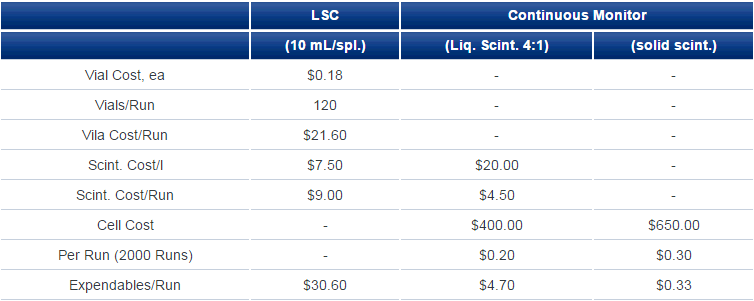
Table 1. Cost Comparisons (Expendables)
There is more to the picture than these costs; there is also performance. With the use of liquid scintillator, continuous-flow H-3 counting at once became practical. With phototubes close together, possible because continuous-flow cells are flat coils, light collection, and therefore counting efficiency, is as good as with a liquid scintillation counter with the same counting mixture. On the other hand, bringing the photomultipliers close together elevates crosstalk -- light interactions between the two of them leading to coincidence counts -- thereby increasing background.
Modern methods
Reawakened by HPLC, interest in continuous radioactivity measurement has encouraged reexamination of methods other than admixing eluate with liquid scintillator though that technique remains most widely practiced, perhaps by a factor of 5:1 over all other methods combined. Anthracene is completely forgotten; other solid scintillators are far more practical (though none has quite as high a photon yield). Cerium-activated scintillating glass, calcium fluoride and yttrium silicate packed in flat coil cells of Teflon tubing (Figure 3) have each found acceptance. H-3 efficiencies are still low and the problem of sequestration of activity by scintillator has by no means been eliminated. Yet, these materials are relatively inert and may be treated more rigorously for decontamination than anthracene. And, of course, they are not affected by organic solvents.
It is difficult to be precise about solid scintillators; one must test them to know what to expect. In general, scintillating glass is the poorest of the three, and the one least used, but also the one least easily contaminated. For analytical work it is useful for C-14 (20-40% efficiency) but not for H-3 (>0.1% efficiency); it is recommended for preparative work where high activity levels obviate the need for high efficiency. 60-100 micron yttrium silicate and calcium fluoride offer more or less equivalent performance (40-60% efficiency for C-14, 1-3% efficiency for H-3) and seem to differ more by batch than inherently. The choice between the two is probably best made according to which least sequesters the materials being counted and/or the pH; yttrium silicate produces substantial spurious light below pH 3.
However, we do find a problem with particle size. Large scintillator particles (60-100 microns has become the de facto standard) mean low backpressure and unrestricted flow while finer particles lead to higher backpressure and ruptured cells. But, the finer the scintillator, the smaller the interstitial volume and the shorter the path between decay site and scintillator surface. The result is less energy loss as a beta-particle traverses the mobile phase from the site of decay to the scintillator surface. This is especially important for H-3 where even the most energetic particle gives up all of its energy while passing through less than 1 mm of water.
There is an obvious solution -- construction of cells that can withstand the backpressures that develop as finer particle sizes are employed. Built of heavy glass, quartz, or even sapphire, some are able to withstand 3000 psi, or even more. They permit use of particles in the 10-30 micron range. While C-14 efficiency is somewhat enhanced, the real advantage is for H-3 where the efficiency may be as high as 10%. Unfortunately, there is a negative aspect; as particle size is reduced total surface area is increased and so is the possibility for contamination. The accepted compromise is the use of 60-100 micron particles which allow good eluate flows -- 1-2 mL/min -- at backpressures of less than 300 psi, whenever possible, and to employ the more costly high pressure cells only when necessary.
HPLC has also made a modified form of Cerenkov counting a practical means for continuous-flow detection. Cerenkov counting involves measurement of light produced by energetic beta-particles moving through a medium of high refractive index faster than the speed of light in that medium; the intensity of light emission is much like H-3 and, in fact, most Cerenkov counting is performed under H-3 counting conditions. In early 1964, when I suggested (9) that a liquid scintillation counter might be used for Cerenkov counting of P-32 (Emax = 1710 kev), my thought was, as Steinberg's had been, that scintillator could be saved and sample recovered. In the coincidence-type liquid scintillation counter a colorless P-32 sample in water can be counted with 20-30% efficiency and various additives, not unlike scintillators, may double the performance (10).
At the time my suggestion reached print, there is no evidence that it was of interest to the few then using continuous-flow. But had Cerenkov counting been tested, it would not have been wholly satisfactory. That was not because it did not work, but because broad peaks brought about by slow chromatography combined with H-3-like efficiencies meant even lower Cerenkov counting efficiency than the 20-30% that can be had in a static counter, very much reduced from the 80-95% that P-32 would have given had the same eluate been counted with either liquid or solid scintillator.
Even though the additives that enhance discrete sample Cerenkov counting are not conveniently used in a continuous system, once HPLC came along to sharpen peaks, the lower efficiencies of Cerenkov counting became acceptable because the signal-to-noise ratio was so greatly improved. More recently, new cell designs, i.e., sandwiching the cell coil between disks of plastic scintillator, have improved performance so that P-32 can be counted with 70-80% efficiency. Energetic particles which pass through and out of the cell coil without causing much Cerenkov effect generate light when they interact with the scintillating plastic. That light is more than sufficient to cause both photomultipliers to pulse. The net effect is to combine Cerenkov counts within the cell coil with scintillation events taking place outside, thus enhancing efficiency. Unfortunately, this method is restricted to energetic isotopes; there is a threshold of 263 kev for the Cerenkov effect in water and probably even more energy is required for a beta-particle to be sufficiently penetrating to pass through the eluate, then the coil walls, air, and finally strongly interact with external plastic scintillator.
Input stream splitter
Addition of liquid scintillator to a chromatography eluate, whether continuously or in test tubes, almost always makes the eluate unsuitable for any purpose other than counting. With fraction collection, when some of the eluate must be saved, it is recovered from the individual collection tubes by pipetting, thereby doubling the number of samples. Then, once all fractions are counted and the location of the activity pinpointed, the samples of interest are segregated and the others discarded. With the continuous monitor, and solid scintillator or especially Cerenkov counting, this can become a nonexistent problem; the eluate leaving the detector passes directly to the fraction collector which is actuated automatically as peaks are detected.
When these counting methods are unsatisfactory and liquid scintillator is required, the continuous-flow detector is provided with an input splitter which precedes the point of scintillator input; some part of the incoming stream is diverted and collected in a fraction collector either on a timed basis or with indexing governed by the activity levels detected from the other part of the stream. Two forms of splitter are in current use, either a tee which provides a fixed split or a fast acting three-way valve operating on a short cycle, typically one second, during which one exit port is open to a fraction collector for a part of the second and the other exit port is open to the continuous-flow detector for the other part.
Most splitters are of the second type, i.e., they employ a valve. This gives greater flexibility since the division of time can be controlled as the situation warrants. It also permits computation of total activity with compensation for the split, the computer entry which controls the valve also being applied as a correction factor to the measured counts. The inherent error in first directing all of the stream one way and then the other, rather than making a true split, apparently does not create a problem as long as the operating cycle of the valve is relatively short as compared to peak width.
Waste disposal
Not only are supply costs less than for fraction collection and discrete sample counting, but a new element -- waste disposal cost -- has been added to the equation. Eliminating low-level radioactive waste is not only costly, but with all but one dump site in the U.S. now closed, there is almost no place to put it. Today, disposal of a 55 gal drum filled with radwaste costs over $1000 whether filled with scintillation vials and their contents or just the liquid waste from perhaps ten times as many continuous-flow runs. It is true that both types of waste can be incinerated but that from a discrete sample counter must first be subjected to crushing, separation of glass and plastic, and these solid wastes must then be treated.
Even beyond this, when solid scintillator (or Cerenkov) counting is employed for continuous-flow measurement, there is essentially no disposal cost.
And to suggest further concern over this point, modern continuous-flow systems often include a waste management device, essentially a three-way valve which responds to radioactivity detection and directs radioactive waste to one collection vessel and non-radioactive waste to another. Depending upon the peak widths of activity present, the volume of radioactive waste may be decreased by 75% or more; it is a rare HPLC run in which radioactivity flows even 10% of the time.
Performance considerations
Not only does the modern continuous-flow monitor offer economy and convenience, but also improved performance and improved data handling. Performance enhancement, apart from that which derives from new cell design and a better choice of scintillators, has come about by virtue of substantially reduced "background" which leads to increased sensitivity. The "background" of a liquid scintillation counter results from ambient radioactivity, against which shielding is effective, K-40 in the counting vial which produces both Cerenkov light in the glass and scintillations within the counting solution and for which shielding is not possible, and other light-producing phenomena within the photomultiplier which give rise to "crosstalk"; electronic noise is no longer a problem while the use of coincidence minimizes the effects of tube noise.
"Crosstalk" is the consequence of spurious photon production in either photomultiplier being seen by both photocathodes and therefore registering a false coincidence. It comes about as trace radioactivity in the photomultiplier envelope generates Cerenkov light and also from collision of internal electrons and ions with the electrode support structure and the envelope. Crosstalk is less a problem with discrete sample counters where phototubes are 35-40 mm apart to accommodate the counting vial than it is with the continuous-flow monitor where there is only a flat coil of tubing and 10-15 mm spacing is used to improve light collection. That crosstalk is more important in one device than in the other is shown by counting empty chambers where, with other factors equal, the count rate of the continuous-flow monitor is three or four times that of the liquid scintillation counter.
But, the crosstalk problem has been overcome, in part through use of appropriate photomultipliers. Our first continuous-flow monitor employed 2" diameter phototubes, the same phototubes used in the liquid scintillation counters we were then making. But, today, circumstances are different, not only because the two-tube spacing is different, but also because of the physical attributes of the sample container. In the one case a vial chosen to facilitate sample preparation, typically 25 mm dia x 50 mm tall, dictates photomultiplier size while in the other case a flat coil can be wound to conform to whatever photocathode configuration is preferable.
Possibly the first to recognize that using the 1" photomultiplier in a continuous-flow monitor was advantageous was H. Kubisiak whose Raytest Strahlungsmessgerate GmbH introduced, in 1983, the Ramona Flow-Through Monitor (Figure 4). Based upon 1" phototubes, the Ramona offered "backgrounds" which were half those of competitive instruments while counting efficiencies were quite the same. The reason was simple; crosstalk was down by perhaps 75%. With a 2" photomultiplier having four times the photocathode surface, four times the base surface, two times the cylindrical wall surface, and four times the volume of a 1" tube, it is no wonder that the 1" tube exhibits so much lower background.
Once it was shown that crosstalk was so important in continuous-flow monitors and that with proper attention to it, backgrounds could be substantially reduced, manufacturers have improved their product. Some do use 1" tubes which offer the additional advantages of lower cost and more compact design (Figure 5), others continue with 2" tubes but employ electronic means of crosstalk suppression. Then, with plastic coil cells which contain no K-40, and with cell volumes of 0.5-2 mL being so much smaller targets for ambient radioactivity than the 10-15 mL of the counting vial, the net result is lower background for the continuous-flow monitor than for the discrete sample counter.
While subjective comparisons to this point favor the continuous monitor over discrete sample counting, what do objective tests show? There is not much question that, when taken to its limits, i.e., lengthy counting times, discrete sample counting offers the possibility of greater sensitivity. Nevertheless, once activity levels are even slightly elevated, differences between the two methods are surprisingly small (10). Table 2 is indicative; the Lower Level of Detection in cpm, calculated according to the expression:
LLD = 4.66 (B/T)½
where:
B = Background, cpm
T = Measurement Time, min
is compared for typical background levels and counting times. It tells us, with 95% confidence, that an observed count rate of LLD is significant, i.e., that there is only a 5% chance of there not being true activity. A superficial glance suggests that discrete sample counting enables one to see lower levels. But, that does not recognize that the backgrounds of the continuous monitor tend to be the lowest used in these calculations while those of the liquid scintillation counter tend to be the highest. Also, as elsewhere suggested, on average, collected peaks will be bisected; for comparative purposes, levels attainable with the continuous-flow system should be examined against twice that level for discrete samples. When these adjustments are made, discrete sample counting remains the more sensitive, but by much less than cursory examination of Table 2 implies.
Another test whose outcome is given in Table 3 is to consider accuracy. Here, we assume a 60-second wide peak, a background of 10 cpm, and 15 seconds of counting time for the continuous monitor and 1 minute for the liquid scintillation counter. At two different confidence levels we compute the activity necessary to achieve a predetermined accuracy with a liquid scintillation counter based on the approximation:
A2 = (K/T) x (1/N + 2B/N2)
where:
B = Background
N = Net Counts
K = l (68% confidence)
K = 4 (95% confidence)
Then, reversing the process, we assume that count level and determine what would be the accuracy at that same count level under continuous-flow conditions. At all but the lowest levels, differences are small.
Then, there is the question of resolution. Certainly if one collected a great many fractions and counted each for a lengthy period, fraction collection must win out. But, if we restrict such a discussion to practicality, unless count rates are so low as to be almost imperceptible, the continuous-flow monitor provides an inherently better opportunity to see elution profiles than does fraction collection followed by liquid scintillation counting. Considering laboratory reality, on the outside the aforementioned 60 minute chromatography run with 120 fractions, and peaks that are 30-seconds wide, how do the two methods compare?
A fair test is to examine two peaks, probably of different amplitudes but each of width t, and separated by a baseline region again of width t (Figure 6). What can we expect when collecting fractions also of width t? Labeling the first tube to contain radioactivity "Tube 1", apart from the highly improbable perfection of Figure 6a, then four consecutive tubes will always contain some radioactivity while the ones on either side will never contain any (Figures 6b-d). On repeat runs the collection pattern is not likely to be quite the same because there are always slight variations in the chromatography which might affect retention times by a few seconds.
Possibilities that meet these conditions are shown in Figures 6c and 6d. On examination, though there is no indication of separation quality, they do suggest two peaks because it seems that there are two maxima separated by one tube with lesser activity. However, suppose that the elution pattern was not known and the collection period had been 2t? We might very well see the results shown in Figure 7. Here, two peaks always appear as one, a totally unacceptable result.
More fractions might clarify the situation but would also divide up the activity necessitating longer counting times. More tubes would further increase supply and disposal cost and increase work in the laboratory; if peak width is 30 seconds and fraction collection time is also 30 seconds -- normal sorts of numbers -- it is usually not practical to collect additional fractions.
What would be the corresponding situation with a flow-through monitor? With the flow-through monitor peaks appear wider than they really are as a result of the substantial transit time through the measurement cell, the width displayed being the sum of true peak width plus transit time (Figure 8). Thus, if transit time should happen to equal peak width, our two peaks will still be evident, albeit with almost no separation as the first peak leaves the cell just when the second enters. With a larger cell, e.g., one having a transit time of 2t, the peaks will appear fused but with a step since for a short while parts of both peaks will be in the cell at the same time. On the other hand, with a cell having a transit time of t/2, the two peaks become clearly evident but the number of counts recorded is reduced.
Here, the continuous-flow system offers obvious advantage. Additionally, results are reproducible; there is none of the randomness associated with the appearance of a peak vs. the actuation of a fraction collector. Whatever the cell size, results are still presented in real-time. More important, it is practical to change cells in a flow-through system so that the transit time is shortened to where peaks don't come together; the cost is only in counts but not in supplies, disposal, equipment utilization, labor, etc.
And even the count reduction must be put into perspective. On average, with true peak width approximating collection time, which is not at all unusual, the fraction collector divides peaks whereas the continuous-flow monitor looks at the entirety. So, when a smaller cell is chosen to improve resolution, the operator is merely moving toward what is normally expected with fraction collection; the fraction collector begins with a diminished starting point and, as a practical matter, that disadvantage is not easily overcome.
External data inputs
Another most useful contribution from Kubisiak was the provision of an analog input channel and an ADC, so that the Ramona monitor could be used to record mass and radioactivity simultaneously. This concept has been expanded so that all modern continuous-flow monitors now provide at least two such analog input channels and some even offer more. These inputs are used to monitor UV at two different wavelengths, to examine the outputs of diode-array and electrochemical detectors, to keep track of gradients, etc.
There is more to this implementation than one might think. Mass detection cells have small volumes -- 5-10 µL is usual -- while radioactivity measurement cells tend to be as large as the required resolution permits, possibly up to 2.5 mL. The consequence, especially with larger radioactivity cells, is a time displacement between the records produced by each detector which makes it inconvenient to view the two or more traces on the same time axis. Kubisiak, provided a clever solution to the problem. The digitized external input was stored second-by-second in a temporary memory and then output only after a suitable delay to bring that record into coincidence with the radioactivity record. The output might be the digitized information or that might be changed back to an analog signal with a D-to-A converter.
Data reduction
Data treatment logically closes the discussion. With personal computers so common, so inexpensive, and so powerful, it oughtn't be surprising to find a computer in some way associated with almost every new continuous monitor and liquid scintillation counter. Because the continuous-flow monitor is meant for a single predictable purpose, whereas the liquid scintillation counter is a general purpose instrument, the software which accompanies a continuous monitor is more focussed and takes the data further toward its ultimate end use. Further, the appearance of data from a continuous radioactivity monitor is not unlike that from other detectors used to monitor HPLC and GC columns for which an abundance of software has been developed.
But, there are also differences: Radioactivity measurement is subject to statistical fluctuations which, especially at low levels, give a ragged and sometimes indecipherable appearance to the data. The nature of the mobile phase, and to a lesser degree the nature of the analyte, sometimes affect the scintillation process to degrade counting efficiency ("quenching"). When measuring two radioisotopes in the same sample, there is always overlap with some contribution of the more energetic isotope appearing in the energy range of the less energetic. Peaks appear broadened both by the relatively large cells which radioactivity measurement requires and also by the addition of liquid scintillator. Proper treatment of radioactivity background requires greater sophistication than does mere baseline subtraction. Should both mass and radioactivity measurements be made during the same chromatography run, the time displacement (as well as a peak width differential) must be taken into account.
These go beyond conventional chromatography data treatment but have been addressed by the manufacturers of continuous-flow radiochromatography instrumentation. Their software is something of a composite. It provides automatic peakfinding and peak integrals, retention times, percentage recoveries, and prints graphs and data tables; it allows permanent data storage and offers protocols which conform with GLP and GALP. And superimposed upon this are data smoothing methods to account for statistical fluctuations, corrections for background, quenching, energy overlap, scintillator dilution, flow rates and cell volumes, etc. Since so complete a treatment is not normally found in the software which accompanies discrete sample counters, the convenience available to users of continuous monitors has been among the more prominent reasons for the growth of this field.
Conclusion
Though at the lowest levels of beta-activity, continuous-flow measurement in HPLC eluates might be marginally less sensitive than would be fraction collection followed by discrete sample counting, that possible lower sensitivity is the only disadvantage that can be cited. Otherwise, every other factor that one might consider -- first cost, supplies, labor, waste disposal, resolution, data treatment, real-time results -- all favor the continuous monitor. The real advances in chromatography that have come about in recent years have combined with improvements in radioactivity measurement to make the continuous-flow monitor the preferred system for radio-HPLC detection.
References
Rapkin, E. and Packard, L.E., U.S.A.E.C. TID-7612, 216 (1960).
Rapkin, E. and Gibbs, J.A., Nature 194, 34 (1962).
Steinberg, D., Anal. Biochem. 1, 23 (1960).
Schram, E. and Lombaert, R., Biochem. J. 66, 21P (1957A).
Schram, E. and Lombaert, R., Anal. Chim. Acta 17, 417 (1957).
Schram, E. and Crokaert, R. Biochem. J. 66, 20P (1957A).
Funt, B.L. and Heatherington, A., Science 129, 1429 (1959).
Scharpenseel, H.W. and Menke, K.H., Tritium in the Physical and Biological Sciences, Vol. 1, p. 281, I.A.E.A. Vienna (1962).
Rapkin, E., Int. J. Appl. Radiation & Isotopes 15 , 69 (1964).
Rapkin, E., J. Liquid Chromatography, 16(8), 1769 (1993).
Elrick, R.H. and Parker, R.P., Int. J. Appl. Radiation & Isotopes 17, 361, (1966).