Modern Tritium Handling in the Synthesis Laboratory
Edward Rapkin, Gavin Steele, and Russell Schavey of IN/US Systems, Inc. As published in American Laboratory, Volume 27, No. 15, p 31 (October 1995)
As with many materials whose potential hazards were long unrecognized, or whose profligate use for so long bore no social stigma, the proper handling of tritium gas in the chemical synthesis laboratory has, until recently, not received adequate attention. Once tritium became freely available as a consequence of both weapons production as well as the increasing number of heavy water reactors, use of tritium labels in the bio-medical laboratory blossomed. Tritium was inexpensive, even cheap, it is conveniently measured with liquid scintillation counters, and, best of all, it is not very energetic and therefore seemingly (and in actuality) rather safe and forgiving.
Tritium (3H / "T") is a pure β -emitter with an Emax of 18 keV. It has a half-life of 12.26 years; its decay product is non-radioactive 3He. Pure tritium gas (T2) has a high specific activity, just over 57,000 Curies/mol. With hydrogen being present in just about every complex organic molecule, tritium is in many ways an ideal label.
The argument for tritium as contrasted with other likely labels is made with the aid of Table 1:
Table 1 - Specific Activity of Selected Nuclides
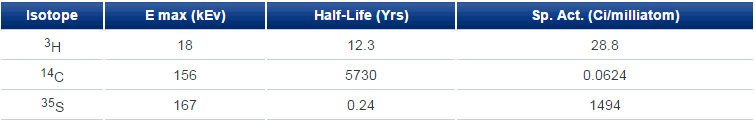
where specific activity is shown for 100% enrichment; 3H has nearly five-hundred times the specific activity of 14C while 35S, with its short half-life, is fifty times more active than 3H. However, the relatively infrequent appearance of sulfur in bio-organic molecules and its perhaps too short half-life makes 35S of less interest than either of the other two.
With increasing interest in high specific activity tritium labeling, there is a correspondingly increasing interest in improved tritium handling. Some of the most useful tritiation techniques involve gaseous tritium either as a direct reactant or in the formation of a tritiation reagent, which sometimes is as simple as water.
Classical laboratory methods of working with tritium gas involve storage and manipulation in glass vacuum lines, moving the gas about with Topler (mercury-piston) pumps. Stopcocks are normally avoided because they leak, sometimes freeze, and because of the possibility of exchange and contamination with stopcock grease. Mercury float valves are better but still an unpleasant necessity with the fragility of a glass line suggesting potential problems from mercury vapor and, of course, from large quantities of gaseous tritium.
While tritium gas has been inexpensive and readily available, that situation may be changing and excess tritium recovery could become important. Normally more tritium must be purchased than is needed for a single preparation and not all that enters the reaction flask is utilized. It is accepted that 80-90% of all tritium which enters into glass lines is ultimately lost or unusable. Long-term storage of large amounts of radioactivity in a glass globe appended to a glass line does not inspire confidence, not only from a safety standpoint but also because as radioactive decay inexorably proceeds, the tritium becomes diluted with 3He which makes for later problems in dispensing known quantities.
None of this can continue. Tritium, is still inexpensive though the longer the Savannah River reactors remain closed, the scarcer it is likely to become. Even, if this were not the case, loss of 80-90% of incoming material to the atmosphere, or even to proper (and costly) disposal is no longer acceptable. The increasing complexity of regulatory procedures also suggests that a repetitive user would best arrange for a higher possession limit (which implies the need for a safer, more reliable system) to bring in tritium only occasionally, and obtain it pure when needed from convenient on-site storage.
Surely, there must be some better way than the conventional glass line. That hydrogen (and deuterium and tritium, too) can be sequestered in large amounts on metal surfaces with subsequent controlled release by heating has long been known. Indeed, much effort, but as yet without commercial success, has been directed to absorbing hydrogen on iron for use as an automotive fuel. For this purpose, iron is chosen over other metals because it has acceptable properties and is low-cost. But, upon examining various metals when cost is not so critical and convenience and safety are, depleted uranium stands out.
Depleted uranium is essentially pure 238U. It is an alpha-emitter (4.20 MeV, 4.15 MeV) with a low-energy (0.05 MeV) gamma component; its half-life is 4.5 x 109 years. Radioactivity is not a problem and is not detected externally when depleted uranium metal is held in almost any container. At mildly elevated temperature (200-300 degrees C) uranium metal reacts stochiometrically with all three hydrogen isotopes to form the trihydride, trideuteride, or tritritide (UX3) (1); the synthesis process is reversed by heating. And, once the process has gone through one complete cycle, the uranium surface is so greatly increased that subsequent conversions to UX3 will occur at even lower temperatures verging on room temperature.
1 g of uranium in the form UX3, equals 1.26 x 10-2 g atoms of hydrogen isotope; as the pure tritide that is 363 Ci. With uranium having a density of 19.05, large quantities of tritium can be stored in a small volume. For example, storing 1 mol of gaseous H2, D2, or T2 in a 100 cc container requires a pressure of 240 atmospheres (3500 p.s.i.) at 20 degrees C; the same amount of isotope is stored on uranium at atmospheric pressure in a total volume of less than 10 cc.
At room temperature, the equilibrium partial pressure of any of the three isotopes above uranium is variously reported to be from 2 x 10-3 Torr to just over 1 x 10-6 Torr, at 200 degrees C it is less than 1 Torr, and it does not reach 760 Torr until 400-450 degrees C. Table 2 (2) is representative of data that can be found in the literature:
Table 2 - Equilibrium Partial Pressure of UT3 vs. Temperature

One byproduct of this process is that it facilitates partial purification of tritium. Tritium gas diluted with the inevitable 3He and/or with nitrogen is easily rid of these contaminants by first forming UT3 and then pumping on it at room temperature. As Table 2 clearly demonstrates, under such conditions and even with large quantities of tritium, there will be no significant tritium activity in the pump exhaust. And, this is a continuing thing; no matter how long the tritium is held as UT3, if pumped on prior to heating, the tritium that is then liberated will always be free of 3He.
Thermal release of absorbed gas from uranium is notable for its smoothness. In a closed system the pressure increase is continuous, following the temperature rise without spurts or jumps; one needn't be concerned with runaway release or overly rapid pressure build-up. Upon cessation of heating, there is almost immediate total reabsorbtion of free tritium. By the time the temperature has dropped to 200 degrees C, which with a relatively small amount of uranium can be a matter of a minute or two, the pressure has dropped to just a few Torr. In another few minutes as the uranium bed continues to cool, the equilibrium partial pressure drops another several orders of magnitude while the observed pressure bottoms the gauge.
We have what seems an ideal situation. Depleted uranium is neither so exotic as to be unavailable nor so expensive as to be prohibitive; it is an article of commerce. It sequesters large amounts of tritium which it releases under relatively easily controlled conditions which are sufficiently different from ambient to be inherently safe. And, when the tritium in UT3 decays, as it must, the decay product is 3He which is non-radioactive and may be pumped off to the atmosphere. Thus, the use of uranium offers the potential for a safe, easily controlled temporary store for tritium; an obvious extension is to conduct tritiation reactions in the same system.
The TRI-SORBER Tritium Manifold is an industrial realization of these ideas, largely based upon the work of Morimoto and Williams (3). It combines the technology of uranium absorbtion of hydrogen isotopes with modern gas handling equipment and methods. An all stainless steel line is used, welded wherever possible to prevent leaks. Packingless, high vacuum bellows valves assert control but do not introduce potential weak points; the valves toggle on and off with a simple lever action to eliminate the risk of damage from overtightening.
A built-in vacuum pump, normally an oil pump, produces an ultimate vacuum of less than 1 x 10-2 Torr. Should disposal of contaminated oil be thought a potential problem, the TRI-SORBER can be supplied with a molecular drag pump and oilless roughing pump capable of even lower ultimate vacuum. And interposed between the pump and the manifold there is an automatic shut-off valve to isolate the system from the outside environment whenever the pump is not operating.
The three uranium beds -- one for fresh tritium, one to recover excess, one for deuterium pilot studies -- are machined from solid stainless bar stock to eliminate potential welds which might be subject to damage from repetitive heating and cooling. The beds are fixed in place and are surrounded by copper heater blocks, each with its own cartridge heater. The blocks are on tracks and are pulled forward to initiate heating and pushed rearward to stop.
During a normal tritiation run, heating stops automatically when a preset gas pressure is reached. With the system having a known volume, presetting a pressure end point allows the operator to liberate that amount of tritium required for the instant reaction. The appropriate quantity is captured in the reaction flask, and before other operations have been performed, excess gas in other parts of the system is returned to the now cooling storage bed. With modern catalysts, most tritiations can be run at sub-atmospheric pressures. This not only conserves tritium but, should the system exhibit minor leakage, gas flow is inward rather than outward and the pressure rise signals the need for extra care.
The TRI-SORBER has been fully instrumented for safety. Proportional heater control governs the rate of heating and also prevents significant temperature over- and undershoot. Gas pressures are continuously monitored and displayed; in addition to the operating pressure preset, a redundant maximum pressure limit (950 Torr) operates a heater cut-out to ensure that excessive gas pressures cannot be generated. There is logical protection against heating more than one uranium bed at a time and/or heating the wrong bed. When the system is not in use, provision has been made to flood it with helium or argon to prevent inward migration of surrounding air.
The TRI-SORBER includes a charcoal-packed stainless steel trap used to remove solvent vapor from excess tritium gas prior to tritium recovery. Sequestering solvent vapor on charcoal rather than allowing it to come in contact with uranium results in a longer life for the second uranium bed for storage of the tritium excess sometimes needed to drive a reaction to completion. Also, this simple clean-up enables the operator to employ recovered tritium for less critical or large-scale preparations rather than having to dispose of it.
But, should there be no interest in using recovered tritium, this second bed serves as a convenient temporary store. A bed containing 3g of uranium can hold over 1000 Ci of tritium. With an annual fill of 100 Ci, the bed will not reach its capacity until fifteen years of use, the annual decay (to 3He which is pumped off to the atmosphere) ultimately approaching the annual fill. At this time when there are essentially no low-level dump sites in the U.S. to which the public has access, many will find this feature of the TRI-SORBER an almost necessary adjunct to their tritium synthesis program.
The TRI-SORBER's two pressure gauges and three heater blocks all have voltage outputs to continuously monitor the entire process from tritium liberation through dispensing, reaction, and the recovery of excess gas. Figure 3 shows a record of heater block temperature, bed temperature, and absolute tritium pressure. Note how smoothly pressure buildup follows the temperature increase until a preset pressure is reached at which point further heating stops. With heating stopped, with one of the beds open to the tritium gas, the pressure drop is remarkably rapid as the uranium again reacts to form the tritide.
On the reaction side, a differential pressure gauge is used for enhanced sensitivity. Figure 4 is a record of the differential gauge output during the course of a reaction. It shows gas takeup as tritiation proceeds; the spike at midrun results from a second admission of tritium. Software has been developed to account for all of the tritium entering the reaction flask, no matter how many additions are made or what the pressure, in order to provide a profile of each run. In this manner, by controlling and following tritium admission, complete or partial reductions can be effected, reaction rates established, and the efficacy of catalysts established.
Finally, it should be stated that the TRI-SORBER, with its all stainless steel construction completely contains all radiation from either the tritium or, as previously noted, the depleted uranium. A system loaded with 200 Ci of tritium showed no measurable external radioactivity either with a monitor or on extensive wipe testing.
Summary
The TRI-SORBER is a complete system for handling tritium gas in the synthesis laboratory. It combines provisions for intake, storage for fresh and recycled tritium and hydrogen or deuterium for pilot studies, means for freeing and metering gas, a vacuum pump, a charcoal trap to aid in recycling, all in a single, compact and inherently mobile apparatus. All of this has been conceived with a keen eye toward safety, not only because there might be hazards in handling tritium but also because the designers and surely the purchasers as well accept the social responsibility which is attendant with working with relatively large amounts of radioactivity.
(1) F.H. Spedding et al., Nucleonics, 7 No. 1, 4 (1949).
(2) H.R. Schultz, in Radioaktive Isotopen in der Organischen Chemie und Biochemie, p. 125, VEB Deutscher Verlag der Wissenschaften, Berlin 1966.
(3) H. Morimoto and P.G. Williams, Fusion Technology, 21, 256 (1992).